어느 특정 단백질이 DNA에 결합하는 위치를 확인하는 '염색질 면역 침전'(Chromatin immunoprecipitation·ChIP)의 데이터 분석을 신속·정확하게 처리하는 SW를 울산과학기술원(UNIST) 연구진이 개발했다.
ChIP는 단백질 결합위치를 조사하는데 널리 사용돼왔다. 특히 특히 엑소뉴클리아제(exonuclease)를 활용한 최신 실험 기술인 ChIP-exo을 통해 고해상도로 결합 부위를 식별할 수 있다.
그러나 특정 단백질이 실제로 DNA에 결합하는 부위인 피크에 대한 판별은 노동집약적 추가 확인단계가 필수적이다. 그래서 대용량 데이터를 신속히 처리하지 못하는 한계가 있었다.

UNIST는 에너지화학공학과 김동혁 교수팀이 이런 문제를 해결하기위해 딥러닝 기반의 ChIP-exo 피크 선별 소프트웨어인 'DEOCSU'를 개발했다고 23일 밝혔다.
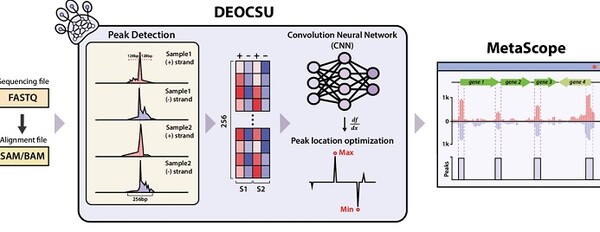
DEOCSU는 참조 서열에 정렬된 ChIP-exo 데이터를 통해 피크 후보를 먼저 감지한다. 이어 각각의 신호를 이미지 데이터로 변환한 후 학습된 데이터를 통해 이미지르 ㄹ작은 단위로 쪼개어 분석하는 기법인 컨볼루션 신경망을 사용해 실제 피크를 선별한다는 것이다.
선별된 각 피크는 위치 최적화와 결합 크기 등을 추정할 수 있다. 해당 결과 데이터는 자체 개발 시각화 소프트웨어인 MetaScope를 통해 확인 가능하다.
DEOCSU 모델은 학습에 사용된 데이터뿐만 아니라 미지의 ChIP-exo 데이터에 대해서도 정확하게 결합 부위를 선별했다.
공개 데이터베이스(EcoCyc와 proChIPdb)의 정보와 선행된 연구 사례를 이용해 기존에 공개된 소프트웨어(ChExMix, MACE, MACE-elite, PeakXus)와 비교했을 때도 우수한 성능을 보였다.
특히, 원핵생물 유래의 ChIP-exo 데이터 뿐 아니라 진핵생물과 고세균 분석에서도 성능이 유지돼 범용성도 확인됐다고 연구팀은 설명했다.
김동혁 에너지화학공학과 교수는 “단백질과 DNA의 상호 작용을 고해상도로 식별할 수 있는 유용한 이점에도 불구하고 분석의 어려움으로 인해 ChIP-exo 실험 기술의 사용이 제한됐었다”며 “이번 DEOCSU의 개발로 분석에 대한 연구자의 부담감을 줄여 관련 연구의 진행 속도를 가속화할 수 있을 것”이라고 말했다.
이번 연구 성과는 생물정보학 연구 권위지인 브리핑스 인 바이오인포메틱스 (Briefings in Bioinformatics)에 1월 25일자로 출판됐다.